Exploiting Aneuploidy in NF1-MPNST: Prognostication and Understanding Pathogenesis

We have demonstrated that MPNST exhibit a high degree of aneuploidy, while their benign PN counterparts are usually diploid. Moreover, leveraging The Cancer Genome Atlas (TCGA) database, we found that Chr8q gain was associated with reduced overall survival (OS) in patients with soft tissue sarcomas, which may explain the poor prognosis of MPNST. We hypothesize that Chr8 gain is a critical driver of MPNST progression, functions by increasing the expression of a set of genes responsible for tumor growth, and correlates with poor overall survival. We aim to define the genes on Chr8q that are essential for MPNST progression; define the dysregulated signaling pathways in cells with Chr8q gain, and determine if the degree of Chr8 gain observed in FISH correlates with OS or development of resistance.
Utilization of Cell-Free DNA Liquid Biopsy for Diagnosis and Monitoring of Tumor Evolution in MPNST
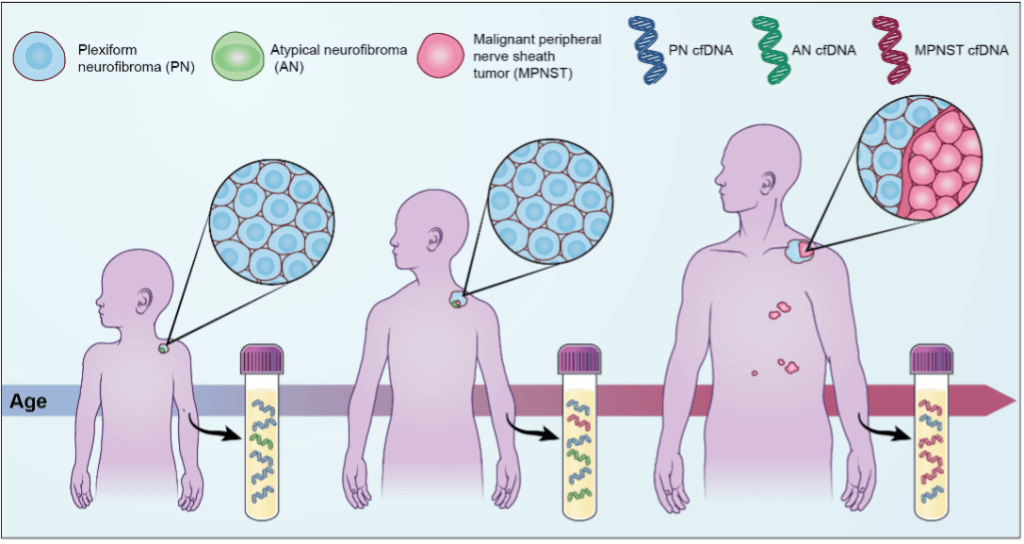
Since MPNST develop from benign PN, the ability to reliably detect malignant transformation is critical to the prompt institution of aggressive management in order to improve survival outcomes. We, and others, have shown that several cancer types can be monitored through plasma cell-free DNA (cfDNA) analysis and that tumor-derived cfDNA is typically shorter in size than normal cfDNA. We have also shown that sequenced MPNST tissue harbors chromosomal copy number alterations (CNAs) that are not present in PN, including in cases of MPNST transformation arising from within benign PN lesions. More recently, we used cfDNA size analysis coupled with cfDNA ultra-low-pass whole genome sequencing (ULP-WGS) to accurately, specifically, and noninvasively distinguish between MPNST and PN patients with 91% specificity and 75% sensitivity. We are actively working to improve this assay through the following specific aims: (1) Design and test a CAPP-seq panel to distinguish MPNST from PN based on alterations in genes and chromosomal regions of biological importance. (2) Integrate CAPP-seq with ULP-WGS applied to cell-free DNA into a unified, ultra-sensitive classifier to noninvasively distinguish MPNST from PN patients. (3) Validate that our integrated CAPP-seq and ULP-WGS liquid biopsy approach can track disease progression and clonal evolution in PDX mouse models.
Regulation of MPNST pathogenesis by the Chromosome 8 gene, UBR5
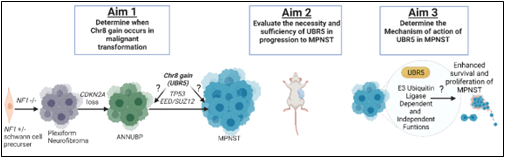
Detailed analysis of Chr8q genes revealed that UBR5 is the most highly upregulated gene in MPNST and that UBR5 genetic knockdown (KD) decreased MPNST proliferation, survival, and migration. Based on these exciting data, we hypothesize thatUBR5 is a key driver of MPNST pathogenesis and in part responsible for Chr8 gain-mediated MPNSTmalignant progression.
TYK2 as a therapeutic target in MPNST
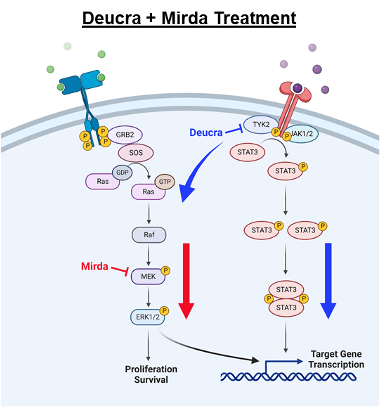
8-13% of individuals with NF1 will develop MPNST during young adulthood. Currently there are few therapeutic options, and the vast majority of people with these cancers will die within 5 years of diagnosis. We previously identified Tyrosine Kinase 2 (TYK2) as a protein overexpressed in the majority of MPNST, whose function is critical for mouse MPNST survival in vitro and in vivo. More recently, we demonstrated that pharmacologic inhibition of TYK2 resulted in a compensatory upregulation of the MEK/ERK pathway, but that combination therapy with a TYK2 inhibitor (deucravacitinib) and a MEK inhibitor (mirdametenib) was highly effective in vitro and in vivo. Current work is aimed at moving this combination forward into clinical trials. Additionally, we are exploring other targets upstream and downstream of TYK2 in MPNST.
Dissecting the Molecular Evolution and Immune Composition in PRC2-retained and PRC2-lost MPNST
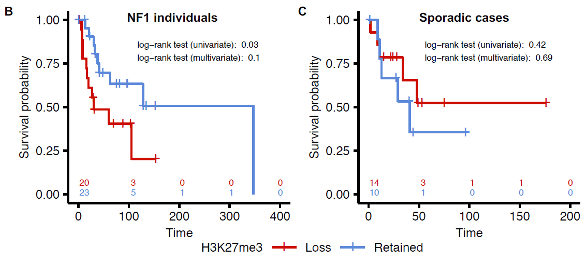
The progression of MPNST is associated with a series of molecular events, including biallelic loss-of-function mutations in NF1, followed by the inactivation of CDKN2A and PRC2. PRC2 is responsible for maintaining transcriptional repression through the catalysis of H3K27me3 via its essential core components, EED and SUZ12. Inactivation of PRC2 leads to the loss of either of these subunits, which in turn leads to the loss of this catalytic activity and in vivo stability. Previous studies indicated that loss of H3K27me3 due to PRC2 inactivation leads to the downregulation of immune response genes, which is associated with lower survival rates and a worse prognosis in patients with NF1-associated MPNST. However, the precise molecular mechanisms underlying the impact of PRC2 loss on the immune profile during progression from PN to MPNST remain unclear. To address this gap, we propose to differentiate the multi-omic profile and immune cell composition of MPNSTs with PRC2 loss and those with retained PRC2 and to determine how tumor evolution differs between these two groups by analyzing PN and MPNST pairs. Finally, in light of the limited therapeutic options available for NF1-MPNST patients, we aim to investigate the potential of epigenetic modifications as a therapeutic target. In our prior studies, we have demonstrated that MPNSTs with H3K27me3 loss exhibit heightened sensitivity to HDAC inhibition with drugs like panobinostat, which modulated immune response pathways. We plan to evaluate the effectiveness of panobinostat therapy in PRC2 loss and retained MPNSTs and determine whether it can augment the effects of checkpoint inhibition-based immunotherapy. This research will provide a deeper understanding of the pathogenesis of MPNST and the immune composition of this cancer, which we hope will lead to a novel, molecularly stratified treatment approach for this deadly disease.
Leveraging a Precision Medicine Platform to Predict Novel Therapies for Malignant Peripheral Nerve Sheath Tumors
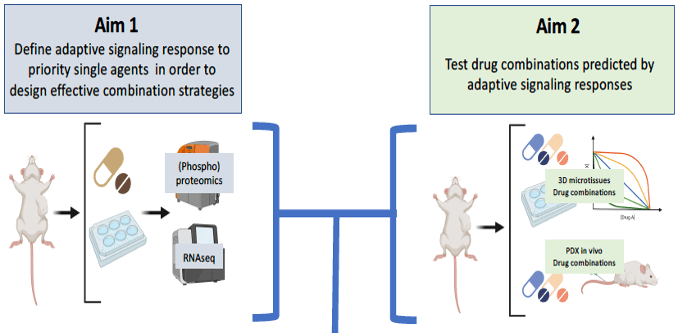
To address the critical need for novel therapeutic advances in MPNST, our labs (WUSTL, UMN, JHU, PNNL) have taken a team science approach to generate in vivo and ex vivo models of MPNST that broadly and accurately recapitulate the genomic diversity of MPNST. This effort has resulted in the largest described set of genomically characterized MPNST patient derived xenografts (PDX) to date, and a unique 3-dimensional (3D) microtissue (MT) culture system in which primary PDX cells and extracellular matrix components are combined and cultured to make a system for medium throughput drug evaluation in genomically-relevant tumor cells These 3D PDX-MT cultures are a platform for rapid, medium-throughput drug studies to prioritize in vivo testing. This system also offers an exciting opportunity to study the adaptive signaling responses to single drugs, test new drug combinations, and find meaningful predictive biomarkers of drug sensitivity in MPNST. The loss of function mutations, and genetic heterogeneity, that characterize MPNST represent challenges for developing effective targeted therapies. In addition to loss of NF1, approximately 60-80% of MPNST exhibit mutations in polycomb repressor complex 2 (PRC2) epigenetic writer genes (biallelic loss of the EED or SUZ12 genes) and these alterations confer a worse overall prognosis. Using high throughput drug screening and other approaches, we have found drugs that are especially effective in MPNST cells with combined NF1/PRC2 loss, including inhibitors of MEK, SHP2, CDK4/6, DNA methyltransferase (DNMT), histone deacetylase (HDAC), and poly ADP-ribose polymerase (PARP), as well as chemotherapies used for MPNST. Moreover, we have identified excellent candidate synergistic drug combinations. Here we plan to continue testing these and other drugs from our ongoing research while also exploiting the signaling changes that occur upon drug treatment to identify agents with the best potential for synergistic activity in vivo.
The Role of ASCT2 and LAT1 as a Diagnostic Markers and Targets in Malignant Peripheral Nerve Sheath Tumors
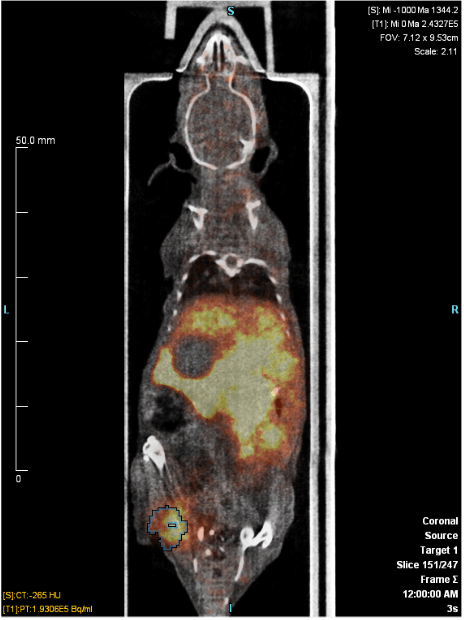
Previous work in our lab led to the identification bIII-spectrinas a protein upregulated in human MPNSTs. bIII-spectrinanchors integral membrane proteins to the plasma membrane as a mechanism for regulating intracellular signaling and cell survival. In this manner, bIII-spectrinhas been hypothesized to control the localization and function of several amino acid receptors and transporters. In exploring the mechanism by which bIII-spectrin affects MPNST pathogenesis, we identified ASCT2 (SLC1A5) and LAT1 (SLC7A5) as amino acid transporters that are expressed in MPNST but not in cell lines derived from benign PN. ASCT2 is a glutamine transporter that is upregulated in a number of different malignancies. Additionally, elevated levels have been shown to correlate with poor prognosis in many cancers including colon, renal, lung, breast, and prostate cancers. Further, ASCT2 serves as a target for diagnostic imaging. 18F-fluciclovine is an FDA-approved non-natural amino acid that is transported by ASCT2 and has been used to image both prostate cancer and gliomas. LAT1, a transporter of essential amino acids has been shown to be expressed in a number of cancers. Additionally, LAT1 expression is associated with higher histological grade and poor survival and LAT1 is a target of an FDA-approved non-natural amino acid tracer, Fluorodopa (18F-FDOPA). We are exploring whether ASCT2 and/or LAT1 can be used as diagnostic biomarkers and may play a role in MPNST pathogenesis.